Vascularite à IgA chez l’adulte, tout sauf un jeu d’enfant…
La vascularite à IgA chez l’adulte n’est pas une mince affaire. Outre les atteintes cutanées et les arthralgies, elle peut entraîner des hémorragies gastro-intestinales et parfois même une atteinte cardiaque. Le facteur pronostique majeur est toutefois l’état des reins, dont la fonction doit être protégée à temps.
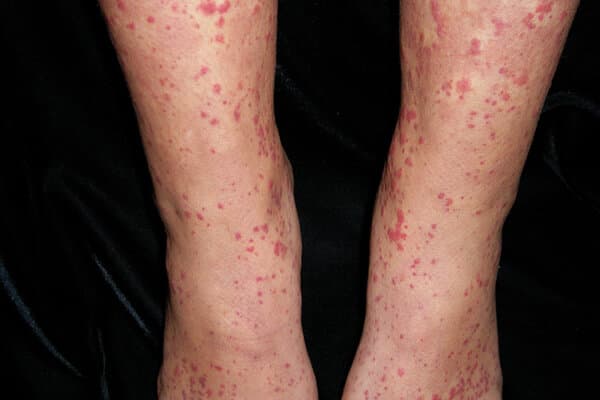
Chez l’enfant, l’évolution de la vascularite à immunoglobulines A (IgA-V), également nommée purpura rhumatoïde ou maladie d’Henoch-Schönlein, est en général bénigne et auto-limitée. Il n’en va pas de même chez l’adulte.
Chez eux, la maladie touche souvent plusieurs systèmes d’organes, récidive souvent et a tendance à la chronicité, écrit le Pr Sabine Adler, médecin-cheffe, service de rhumatologie et d’immunologie, hôpital cantonal d’Aarau. Les hommes sont plus touchés que les femmes, avec un pic de fréquence entre 40 et 50 ans.
Gd-IgA1 se forme probablement secondairement aux infections
La vascularite est due à une glycosylation aberrante de l’immunoglobuline A1 avec production d’IgA1 déficiente en galactose (Gd-IgA1). Cette réaction libère des sites de liaison pour les auto-anticorps pour la formation de complexes immuns. En effet, ces IgA anormalement glycosilées sont à l’origine de la formation de complexes immuns composés d’IgA, de son récepteur CD89 et d’IgG anti-IgA2.
La formation de Gd-IgA1 est probablement la conséquence d’une altération de l’immunité muqueuse, p. ex. secondaire à des infections des voies respiratoires supérieures ou du tractus gastro-intestinal. Enfin, la formation de complexes immuns entraîne des dépôts de complexes immuns et une activation des granulocytes neutrophiles dans les petits vaisseaux. Ces processus inflammatoires peuvent toucher différents systèmes d’organes avec des manifestations variées.
En résumé, l’IgA-V se caractérise par une atteinte inflammatoire avec dépôts d’IgA dans les vaisseaux de petit calibre. Elle évolue par poussées et se manifeste par des atteintes cutanée (purpura vasculaire), articulaire, digestive et/ou rénale. Son pronostic est fonction des atteintes gastro-intestinales et des éventuelles lésions rénales.
L’IgAV se manifeste principalement par une atteinte cutanée (purpura vasculaire) avec un purpura palpable avec formation d’œdèmes sous-cutanés, prédominants aux membres inférieurs. Ils sont présents chez 70 % des patients au stade initial et chez près de 100 % au stade tardif. La présence d’un purpura localisé au-dessus de la hanche signe une atteinte généralisée, associée à un risque accru d’évolution sévère, y compris une atteinte gastro-intestinale et rénale.
Une atteinte digestive dans un cas sur deux
Deux tiers des patients présentent des arthralgies, en particulier des genoux et des chevilles. Les tuméfactions sont fréquentes, mais malgré le risque de provoquer des synovites, l’IgAV n’entraîne pas d’arthrites destructrices.
Des troubles gastro-intestinaux sont observés chez 50 à 80 % des patients, principalement sous forme de douleurs spasmodiques. Des hémorragies ne sont pas à exclure. Elles peuvent aller de formes légères avec un peu de sang dans les selles à des hémorragies mettant le pronostic vital en jeu. Des investigations diagnostiques rapides sont essentielles, notamment pour exclure des perforations ou des obstructions. L’échographie et le scanner montrent souvent des épaississements de la paroi intestinale, tandis que l’endoscopie met en évidence un érythème muqueux et des hémorragies pétéchiales.
L’atteinte rénale touche environ 70 % des patients adultes. Toutes les manifestations sont possibles, de la microhématurie asymptomatique à l’insuffisance terminale. Outre le dosage de la créatinine sérique et le diagnostic de la (micro-)albuminurie, le sédiment urinaire est pertinent. La présence d’érythrocytes glomérulaires signe une vascularite rénale. Les avis divergent quant à la nécessité d’une biopsie. Selon le Pr Adler, son indication est posée au plus tard en cas d’altération de la fonction rénale. Histologiquement, elle se présente en général principalement sous forme de dépôt de complexe immun, comme dans les autres néphropathies à IgA.
Les atteintes cardiaques sont possibles mais rares.
- Des palpitations,
- une tachycardie,
- des épanchements péricardiques et
- des myocardites
ont notamment été décrits. On suppose qu’une vascularite coronarienne est à l’origine de la physiopathologie. En cas de survenue de symptômes cardiopulmonaires, ces signes et symptômes doivent absolument être recherchés. En l’absence de symptômes correspondants, les investigations cardiaques ne se font pas de routine.
Chez l’adulte, un purpura de cause indéterminée est suspect
Le diagnostic de l’IgAV se fonde sur l’anamnèse et la constellation clinique. Chez l’adulte, la présence d’un purpura d’origine indéterminée doit faire évoquer un tel diagnostic. Dans ce cas, une biopsie cutanée peut aider à distinguer la maladie des autres vascularites. L’examen direct par immunofluorescence permet de mettre en évidence les dépôts d’IgA, tandis que l’histologie révèle en général une vascularite leucocytoclasique.
Des marqueurs sérologiques fiables font défaut, écrit la spécialiste. Chez la moitié des personnes atteintes, les IgA – qui donnent pourtant leur nom à la maladie – ne sont pas non plus détectées dans le sang et n’ont donc pas de pertinence diagnostique. Une éventuelle baisse des facteurs du complément n’est certes pas spécifique, mais permet de la distinguer des vascularites des petits vaisseaux.
En l’absence de critères de classification chez l’adulte, on se fonde sur ceux des enfants (voir encadré). Selon le Pr Adler, le critère « insuffisance rénale » complique le diagnostic précoce, raison pour laquelle elle le considère d’un œil critique. Selon elle, il serait préférable d’utiliser le critère « urine : érythrocytes glomérulaires plus/moins albuminurie anormale ».
En l’absence de données claires sur le traitement optimal des IgAV, les recommandations thérapeutiques reposent sur des avis d’experts. Si l'’évolution est bénigne, le traitement symptomatique du purpura et des arthralgies peut suffire. En ce qui concerne les douleurs, il faut préférer le métamizole ou le paracétamol aux AINS.
Traiter les symptômes suffit souvent en cas d’évolution bénigne
Sinon, le traitement est fonction du stade. En cas de protéinurie persistante, les bloqueurs du SRAA et les inhibiteurs du SGLT2 sont recommandés. Le traitement de l’atteinte rénale fait appel aux corticoïdes (par voie orale ou i.v.), à l’azathioprine ou au mycophénolate mofétil (MMF) en fonction de sa sévérité.
Le cyclophosphamide est une option en cas de glomérulonéphrite rapidement progressive. Chez 22 adultes ayant une maladie réfractaire au traitement, le rituximab a également montré une efficacité sur l’atteinte rénale.
En général, l’IgAV évolue souvent de manière intermittente et récidivante, souligne la spécialiste. Dans un collectif de 260 patients dont l’expression de la maladie était principalement modérée, une récidive a été observée chez 15 % d’entre eux, en particulier en l’absence d’une corticothérapie comme traitement de fond. Dans cette cohorte, la survie globale était de 78 % à cinq ans et de 65 % à dix ans.
Au cours de l’évolution de l’IgAV, une atteinte rénale peut survenir avec un certain retard. Pour la détecter précocement, un suivi médical au long cours est nécessaire – même si les patients n’ont (initialement) que des manifestations cutanées ou rhumatologiques légères. En effet, le pronostic de l’IgAV est principalement déterminé par les reins. Jusqu’à présent, l’évolution vers une insuffisance organique terminale était fréquente – toutefois, les experts espèrent que l’introduction de l’inhibition combinée du SRAA/SGLT2 permettra d’améliorer le pronostic rénal également dans le cas de l’IgAV.
Critères EULAR*, PRINTO et PRES pour les IgAV
Les critères diagnostiques des IgAV valent pour l’enfant. Chez l’adulte, ils n’ont jamais été validés à ce jour mais ils ont fait leurs preuves dans la pratique. L’IgAV est considérée comme probable en présence d’un purpura typique plus au moins un autre critère :
- douleurs abdominales ;
- dépôts d’IgA cutanées et rénaux à l’histologie ;
- arthrite/arthralgies ;
- insuffisance rénale.
* EULAR : Alliance européenne des associations de rhumatologie, PRINTO : Société européenne de rhumatologie pédiatrique, PreS : Paediatric Rheumatology International Trials Organisation
- Adler S. Immunglobulin-A-Vaskulitis [Immunoglobulin A vasculitis]. Inn Med (Heidelb). 2024 Feb;65(2):114-121. German. doi: 10.1007/s00108-023-01650-7